
SARS-CoV-2 Variants Detection Using TaqMan SARS-CoV-2 Mutation Panel Molecular Genotyping Assays
- Dec 27, 2021
- 18 min read
Updated: Dec 28, 2021
Infect Drug Resist 2021; 14: 4471-4479.
Published online 2021 Oct 27. DOI: 10.2147/IDR.S335583
Puja Neopane, Jerome Nypaver, Rojeet Shrestha, and Safedin Sajo Beqaj
Patients Choice Laboratories
Copyright © 2021 Neopane et al.
This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution – Non Commercial (unported, v3.0) License (http://creativecommons.org/licenses/by-nc/3.0/). By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms (https://www.dovepress.com/terms.php).
Abstract
Purpose For rapid detection and tracking of SARS-CoV-2, a simple screening method alternative to laborious and expensive sequencing is highly desirable. Here, we evaluated performance characteristics of TaqMan SARS-CoV-2 mutation panel genotyping molecular assay for detection of most common reported SARS-CoV-2 variants using specific RT-PCR assays targeting single nucleotide polymorphisms (SNP).
Patients and Methods A total of 150 SARS-CoV-2 positive samples from March to July were included for this study. In addition, five controls comprised of synthetic RNA B.1.1.7_601443, B.1.351_678597, P.1_792683, B.1.617.1_1662307 and MN908947.3-Wuhan-hu-1 from Twist bioscience and B.1.1.7 (England/204820464/2020) and B.1.351 (South Africa/KRISP-K005325/2020) from Zeptometrix, NY, USA were used for validation. Total RNA from specimens was extracted using Omega Bio-Tek Mag-Bind Viral RNA Xpress Extraction Kit and tested for known SARS-CoV2 variants using ThermoFisher TaqMan SARS-CoV-2 mutation panel molecular assay on the QuantStudio 12K Flex. Nine representative samples have been compared with sequencing. Data were analyzed by genotype calling using QuantStudio™ design and analysis software v 2.5 with the genotyping analysis module.
Results All validation controls were tested in triplicate and repeated in singlet on three different days and all reported variants were matched as expected. Out of 150 SARS-CoV-2 positive specimens, 69 (46%) were B.1.617.2, 49 (32.7%) were B.1.1.7, P.1 and P.2 were 4 (2.7%) each and B.1.351 and B.1.427/B.1429 were 2 (1.3%) each. Three (2%) were B.1.526, and 17 (11.3%) have a mutation in D614G. Genotyping results from the present study showing B.1.617.2, B.1.1.7, and B.1.526 variants and their mutation genes were concordant with sequencing results.
Conclusion Our study indicates that TaqMan SARS-CoV-2 mutation panel molecular genotyping assays detect and differentiate all published common variants B.1.617.2 (Delta), B.1.1.7 (Alpha), B.1.526 (Iota), B.1.351 (Beta), P.1 (Gamma), P.2 (Zeta), B.1.617.1 (Kappa) and B.1.427/B.1.429 (Epsilon) that can be used for surveillance and epidemic control and prevention.
Keywords: SARS-CoV-2, variants, delta, alpha, TaqMan genotyping assays
Introduction
The SARS-CoV-2 virus has constantly changed over time through mutation, resulting in new variants. Several variants of the SARS-CoV-2 have emerged and spread globally during this COVID pandemic.1 Based on the evidence of increased transmissibility, disease severity and reduced therapeutic effectiveness, the Centers for Disease Control and Prevention (CDC) have defined three different levels of threat associated with variants. These include variants of interest (VOI): Iota (B.1.526), Eta (B.1.525), Kappa (B.1.617.1), B.1.617.3, Epsilon (B.1.427/B.1.429), Lambda (C.37) and Zeta (P.2); variants of concern (VOC): Alpha (B.1.1.7), Beta (B.1.351), Gamma (P.1), Delta (B.1.617.2) and variants of high consequence.1,2 Currently, SARS-CoV-2 lineages have been determined by whole-genome sequencing (WGS) of positive SARS-CoV-2 samples which detects new emerging variants and any new changes in viral genomes.3,4 WGS is considered as the gold standard method of identifying new circulating SARS-CoV-2 variants worldwide. However, limited availability made it unfeasible for timely detection of variants for public health response such as contact tracing and determining prevalence of variants of concern in the community.5 Moreover, WGS requires relatively expensive resources and equipment, technical expertise, and a longer time for data processing for sequencing with no established standardized quality control measures. Most of the laboratories around the world do not have the resources required to develop and support large-scale sequencing. At the moment, it is also difficult to implement the WGS in the diagnostic laboratories highlighting the importance of a complementary surveillance strategy. Therefore, for early detection and tracking the spread of particular strains, alternative methods for screening in a few hours is desirable. In addition, rapid and efficient methods for tracking the transmission of certain lineages could be more valuable in situations where mutations are associated with increased transmission, severity of disease, or vaccine failure. With the emergence of COVID 19, most of the laboratories offering nucleic acid amplification tests (NAATs) have the necessary equipment and trained personnel to perform nucleic acid extraction and real-time PCR. Hence, simple genotyping assays using TaqMan SARS-CoV-2 mutation panel may be helpful to rapidly screen for variants and help in surveillance and control of the SARS-CoV-2 infection. TaqMan SARS-CoV-2 mutation panel assays can monitor one or many SARS-CoV-2 mutations by a process that combines TaqMan SNP genotyping technology with a one-step real-time PCR reaction to detect mutations in a sample rapidly and efficiently from extracted RNA. Therefore, we have evaluated the performance characteristics of the Thermo Fisher TaqMan SARS-CoV-2 mutation panel molecular genotyping assay6 for detection and identifying the most common variants (B.1.526, B.1.525, B.1.617, B.1.617.1, B.1.617.3, P.1, B.1.1.7, B.1.351, P.2, B.1.427, B.1.429 and B.1.617.2) using specific RT-PCR assays targeting single nucleotide polymorphisms (SNP).
Materials and Methods
Samples for Validation Seven known validation control samples that include VOC and VOI were used to validate the assay. Five of these controls were synthetic RNA containing SARS-CoV2 variants B.1.1.7_601443, B.1.351_678597, P.1_792683, B.1.617.1_1662307, and MN908947.3-Wuhan-hu-1 from Twist Bioscience, CA, USA, and two of the controls were culture fluids of known SARS-CoV-2 variants: B.1.1.7 (England/204820464/2020) and B.1.351 (South Africa/KRISP-K005325/2020) from Zeptometrix, NY, USA. Total 5×104 copies/μL of synthetic RNA from each control were used to assess the threshold cycle (Ct value) of SARS-CoV-2. From culture fluids, RNA was extracted and determined the Ct value of SARS-CoV-2 using TaqPath COVID-19 combo kits (Applied Biosystem, Thermo Fisher Scientific). All the control variants with Ct value <30 were used for SARS-CoV-2 mutations detection using TaqMan SARS-CoV-2 mutation panel (Applied Biosystem, Thermo Fisher Scientific).
Patient’s Samples Total 150 positive SARS-CoV-2 nasopharyngeal specimens collected from March to July were included and a retrospective study was conducted in accordance with the Declaration of Helsinki in Patients Choice Laboratories, IN, USA. The study was approved by Sterling Institutional Review Board (IRB ID-9190). Informed consent was taken from the study participants during the collection of the samples. Total RNA from all specimens was extracted using Omega Bio-Tek Mag-Bind Viral RNA Xpress Extraction Kit (GA, USA) on the automated liquid handling Hamilton STAR. Ct value >30 were excluded from the study.
Concordance with Sequencing Nine qRT-PCR positive SARS-CoV-2 samples were randomly selected from 150 positive SARS-CoV-2 nasopharyngeal specimens. Total RNA was isolated and determined SNP genotyping using TaqMan SARS-CoV-2 mutation panel. Those nine qRT-PCR positive SARS-CoV-2 samples were sequenced for comparison with genotyping SARS-CoV-2 mutations detection data.
Assay Description TaqMan SNP genotyping assays contain sequence-specific forward and reverse primers to amplify the target sequence region, FAM dye-labeled probe to detect the mutation sequence, VIC dye-labeled probe to detect the reference sequence, and two TaqMan minor groove binder (MGB) probes with nonfluorescent quenchers (NFQ). The one-step RT-PCR takes one hour and 10 min from extracted RNA to results. We used 14 different TaqMan SNP genotyping assays related with S genes and ORF8 gene. They are: S.K417T.AAG.ACG, S.D614G.GAT.GGT, S.E484K.GAA.AAA, S.E484Q.GAA.CAA, S.K417N.AAG.AAT, S.L452R.CTG.CGG, S.N501Y.AAT.TAT, S.P681R.CCT.CGT, S.T20N.ACC.AAC, S.delH69V70, S.delL242.244L, orf8.Q27ST.CAA.TAA, S.A701V.GCA.GTA and S.T478K.ACA.AAA (Applied Biosystem, Thermo Fisher Scientific) with gene name, mutation present and reference and mutant codons.
Genotyping The SNP genotyping along with one-step real-time PCR, of validation controls and patient’s positive SARS-CoV-2 samples were determined using TaqMan SARS-CoV-2 mutation panel (Applied Biosystem, Thermo Fisher Scientific). The tests were performed as per the manufacturer’s instructions. In brief, each one step TaqMan SARS-CoV-2 mutation panel genotyping was performed using 2.5 μL RNA, 0.25 μL TaqMan SARS-CoV-2 mutation panel assay, and 2.5 μL TaqPath 1-Step RT-qPCR Master Mix, CG in 384 well plates with 10 μL per reaction volume. The one-step real-time PCR that combines reverse transcription and DNA amplification reaction was performed in QuantStudio 12K Flex under the manufacturer’s instructions. All validation controls were tested in triplicate and repeated in singlet on three different days.
Data Analysis Genotype calling was performed using QuantStudio™ Design and Analysis Software v 2.5 with the Genotyping Analysis Module. To confer the precision study of the controls, mean, standard deviation (SD), and coefficient of variance (CV) of cycle threshold (Ct) or Cq were calculated. SPSS software (version 17; SPSS Inc., Chicago, IL, USA) was used for data analysis. Chi-squared test was used for descriptive analysis of Supplementary Materials S2and S3. A p-value of <0.05 was considered statistically significant.
Interpretation Based on the allelic discrimination plot, the reference allele that clusters along the X-axis (allele 1 VIC dye) is represented as homozygous allele1, and the mutant allele that clusters along the Y-axis (allele 2 FAM dye) is represented as homozygous allele 2. These cluster plots show very clear discrimination between the wild-type samples and the mutation samples. Based on the mutations identified using 14 different TaqMan SARS-CoV-2 mutation panel assays, VOC and VOI can be detected (Supplementary Material S1).
Results
Validation of Controls
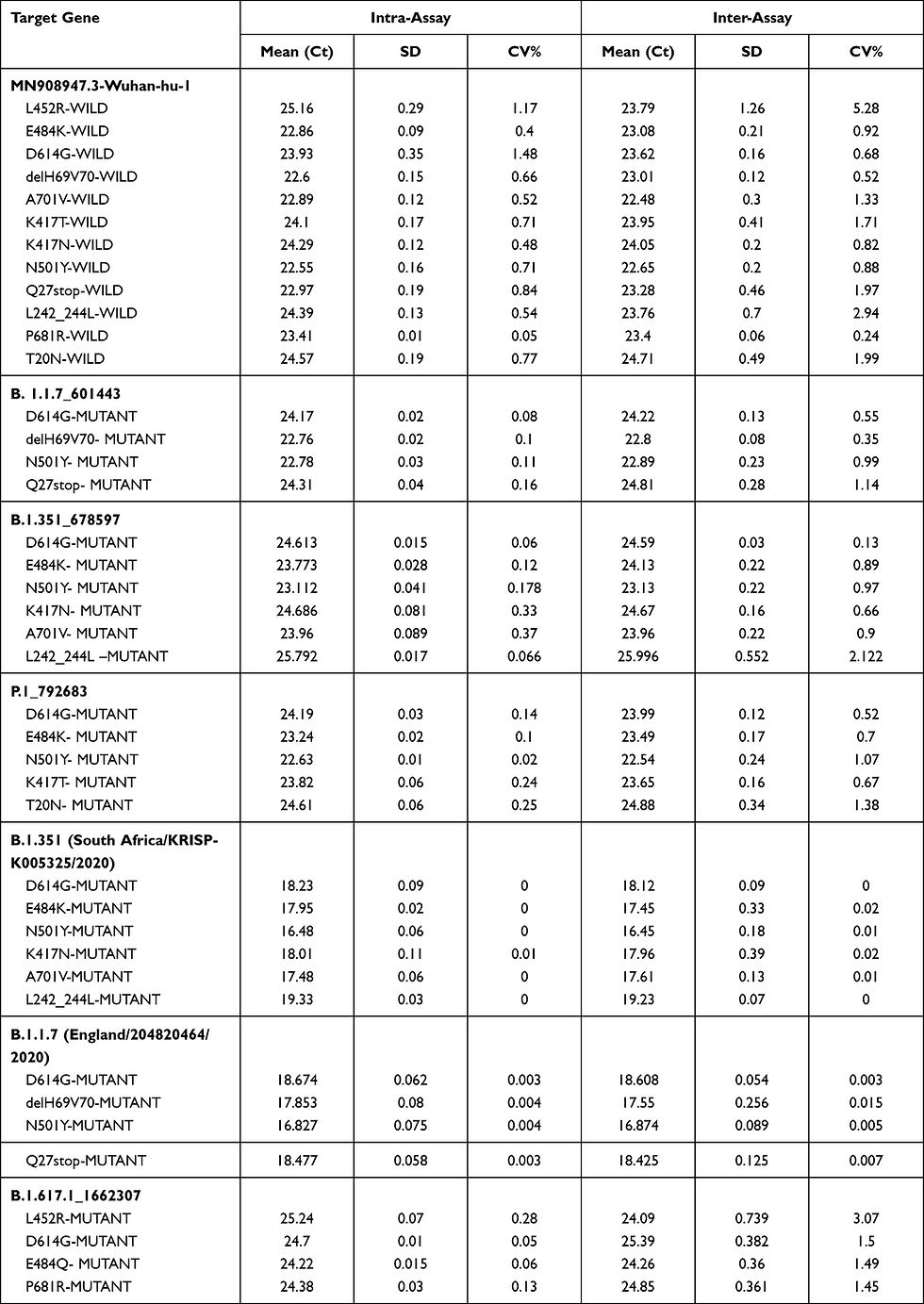
All validation controls were tested in triplicate and repeated in singlet on three different days and all reported variants were matching as expected. The B.1.351 control showed mutation in E484K, D614G, A701V, K417N, N501Y, and L242_244L, B.1.1.7 showed mutations in D614G, delH69V70, N501Y, and Q27stop, the control B.1.617.1 showed mutation in E484Q, P681R, L452R, D614G, and P.1 showed mutations in E484K, D614G, K417T, N501Y, and T20N. The Wuhan-hu-1 did not show any mutation matching expected results, as this control is a reference strain that does not have any of these mutations. The SNP calling result shows 100% agreement with the known expected value from these controls. The precisions study of controls namely, B.1.1.7_601443, B.1.351_678597, P.1_792683, B.1.617.1_1662307, B.1.1.7 (England/204820464/2020), B.1.351 (South Africa/KRISP-K005325/2020), and MN908947.3-Wuhan-hu-1, by intra-assay and inter-assay run showed good reproducibility and repeatability with a CV less than 10%
(Table 1).
Genotyping Results from Patient’s Samples
Demographic Distribution Total 150 SARS-CoV-2 positive samples collected from March to July include 78 (52%) from females and 72 (48%) from males (Supplementary Material S2). There were no significant differences between total of males and females in each month and in all total five months. The total cases of SARS-CoV-2 variants were significantly highest in July (39.4%) than in other months (p<0.05). The number of SARS-CoV-2 variants was significantly higher in the 16–35 years age group than 0–15 years and >60 years (p<0.05) but no significant difference with age group 36–60 years. Cases of SARS-CoV-2 variants are significantly lower in age group 0–15 years than other all age groups (p<0.05). From the age group >60 years, the number of SARS-CoV-2 variants were significantly higher in July with 24 (72.7%) than all other months (p<0.05 (Supplementary Material S3). According to locations, most of the variants identified were from Indiana, Kentucky, and Florida and few were from other states (Supplementary Material S4).
Out of 150 SARS-CoV-2 positive specimens, 69 (46%) were B.1.617.2 (Delta), 49 (32.7%) were B.1.1.7, 4 (2.7%) were P.1, 4 (2.7%) were P.2, 3 (2%) were B.1.526, 2 (1.3%) were B.1.351, 2 (1.3%) were B.1.427, and 17 (11.3%) had mutation only in D614G (Figure 1). Those specimens were from March to July 2021, indicating that these variants were present in-patient population during this time. B.1.617.2 (Delta) was identified from May (23%), 58.8% in June, and reached 94.9% in July. B.1.1.7 (Alpha) variant was highest in April (84.7%), 69.3% in May, 34.3% in March, 17.7% in June, and 5.1% in July. From March SARS-CoV-2 positive samples, 48% of them have only D614G mutation, which did not find in other months. P.2 (Zeta) was detected as 9.3% in March and 3.8% in April. B.1.427 (Epsilon) was detected in 2.8% from March and 3.8% from April. 2.8% of them from March and 11.7% from June were B.1.526 (Iota). P.1 (Gamma) was 2.8% from March, 7.7% from April, and 5.9% from June. B.1.351 (Beta) were 7.7% from May and 5.9% from June (Figure 1). B.1.617.2 (Delta) was significantly highest than P.1, P.2, Iota, and Beta whereas no significant difference with Alpha in total five months (March–July). We determined 35 SARS-CoV-2 negative nasopharyngeal specimens to verify the specificity of the assay used. All SARS-CoV-2 negative samples were unamplified, and no false-positive genotyping signal was detected.

Concordance of SNV Genotyping with Sequencing
All SNV genotyping using TaqMan mutation assay panel results were concordant with the sequencing data. Out of nine samples, five were B.1.617.2 (Delta) and concordant with sequencing showing mutation in D614G, L452R, P681R, and T478K, two samples were B.1.1.7 (Alpha) showing mutation in D614G, N501Y, delH69V70, Q27stop, and two were B.1.526 (Iota) showing mutation in L452R and D614G (Supplementary Material S5).
Discussion
Our validated TaqMan SARS-CoV-2 mutation panel molecular genotyping method can detect the most common and preexisting variants that can help in surveillance and control of the COVID-19 infection. We identified eight different SARS-CoV-2 variants among 150 qRT-PCR positive SARS-CoV-
2 samples that were collected from March to July. The present method requires less than two hours to test after RNA is extracted. Moreover, this test can detect more than 10 variants based on the specific mutation using 14 TaqMan assays. Hence, this method can be a helpful tool for detecting various variants circulating in the community at low cost and less time than sequencing. There are other rapid methods that have been described such as multiplex RT-PCR that includes S-gene target failure, real-time PCR platforms based on melting curve analysis that can differentiate VOC, and rapid antigen tests that may detect the presence of SARS-CoV-2 variant but cannot differentiate the type of VOC.7–10 We detected VOCs and VOI, those having mutation in spike genes, and ORF 8 using TaqMan mutation panel molecular genotyping assays. The present study is in agreement with the previous study that described TaqMan assays as a useful tool for detecting the variants from SARS-CoV-2-positive PCR samples.11 Genotyping data concordance with sequencing data of mutation in the present study indicates that TaqMan mutation panel molecular genotyping assays helps to track the known mutations to screen for different variants.
According to the CDC, the spike gene is the key region for monitoring mutations. These mutations may be related to the increased disease transmissibility, and chances of decreased therapeutic and vaccine efficacy.1 It is noted that the highly mutable spike (S) protein mediates the virus attachment to human cell surface angiotensin-converting enzyme 2 (ACE2) receptor, thus facilitating viral entry during infection.12 The increase in affinity can escalate the host tissue infection, severity of disease and increases the viral load in the upper respiratory tract thereby increasing human-to-human transmission rate.1 The mutations in the D614G, E484K, N501Y, L452R, P681R, and E484Q are associated with the enhanced transmissibility due to increased affinity with host’s ACE2 receptor.12 Deletion of Δ69–70 increases the binding affinity of receptor-binding domain (RBD) for ACE2.12 In addition, E484K disrupts antibody neutralization.1 Although K417N/T mutations decrease the affinity of RBD for ACE2, they facilitate the immune escape.13 The data collected from UK Biobank identified 216 different verified super-variants that were significantly associated with an increase in COVID-19 mortality.14
For early detection and prevalence calculation of known variants, the present method can be a valuable alternative tool to generate results in a few hours. Our finding is consistent with other studies that show a similar rate of SARS-CoV-2 infection in both sexes from epidemiological data.15 Other studies added that the severity of infection was higher in males with a 77% higher rate of mortality.16 Present study found that SARS-CoV-2 was significantly higher in July which was also significantly higher in age group >60 than age group 0–15. This may indicate a higher risk of mortality rates since COVID-19 mortality rates were strongly associated with older age. Age-related dysregulation of ACE2 might be the reason for higher risk factors of COVID-19 complications.17
Our data from March showed that most of the SARS-CoV-2 had mutation particularly in D614G that may be D614G variant previously described.3 The emergence of new variants was led by the global rise of the D614G mutation in the Wuhan strain identified in China and Germany. After March 2020, the D614G clade became increasingly prevalent worldwide, expanding from 22 to 42 countries with GH (D614G + Q57H in the NS3) dominant in North America (59.0%).18 These variants were found to be more transmissible and infectious. Thereafter, many ongoing studies are focusing on new variants that emerged in every country.
We found that B.1.1.7 variant was prevalent in all five months with predominant in April and May. This variant was first identified in September 2020 in the UK and December 2020 in the US.19 Since then, the variant can be found in numerous countries worldwide. Our study found B.1.1.7 variant with mutation mainly in D614G, N501Y and del 69–70 was prevalent in all five months predominantly in April and May. The high binding affinity of these spike proteins for the ACE receptors are associated with increased transmissibility, ranging from 43% to 90% higher compared to the Wuhan strain and/or can partially escape neutralization than other previously identified SARS-CoV-2 variants.19 Furthermore, the study has found that the B.1.1.7 variant was also associated with increased mortality compared to other SARS-CoV-2 variants.20 This is probably the result of an increased number of open RBDs due to the D614G mutation combined with the N501Y replacement in the hAce2-binding site.21 In fact, the B.1.1.7 strain became the most common US strain at the beginning of 2021. In the meantime, many public health strategies were extended such as vaccination, physical distancing, use of masks, hand hygiene, isolation, and quarantine, including genomic surveillance to reduce transmission limiting the spread of SARS-CoV-2. These public health measures might be the reason that the present study exhibited a decline of SARS-CoV-2 variants after March.
We found the B.1.617.2 (Delta) variant from May that reached highest in July (94.9%). The B.1.617.2 (Delta) variant of SARS-CoV-2 was identified in India in late 2020 and has subsequently been detected in approximately 60 countries, including the US.1 Our finding of the prevalence of Delta from more than three states indicates that Delta virus infection was widely spread in various regions of the USA in July. In fact, the CDC also declared at the end of July that B.1.617.2 (Delta) was responsible for more than 83% of COVID-19 cases that were reported in the US.22 It has L452R, T478K, and P681R substitutions in the S protein. The study identified that spike mutation P681R is a significant determinant for enhanced viral replication fitness of the Delta compared to the Alpha variant.23 The effect of the mutations L452R and T478K on ACE2 binding was also observed as enhanced stabilization of the RBD–ACE2 complex. Our result showing gradual decrease in cases of Alpha and increase in cases of Delta from June and July is consistent with a preprint posted in July, which explained that Delta is 40–60% more transmissible than Alpha and almost twice as transmissible as the original Wuhan strain of SARS-CoV-2.24 The same study added that viral loads in Delta infections were 1260 times higher than those in people infected with the original strain.
The recent outbreak of B.1.617.2 (Delta) variant in the gymnastics facility of Oklahoma found the B.1.617.2 variant to be highly transmissible in indoor sports settings and within households.21 The finding emphasized multicomponent prevention strategies including vaccination to reduce the spread of SARS-CoV-2. It was estimated that hospitalization is twice as likely in unvaccinated individuals with Delta than in unvaccinated individuals with Alpha.25 Furthermore, due to only 48% of the total US population having been fully vaccinated, chances of increasing and spreading of SARS-CoV-2 is predominant.24 According to the CDC, in July 2021, within one week, the average increase in new COVID-19 cases and hospitalizations were 69.3% and 35%, respectively.26
Our study presented the highest Delta variant cases among age group >60 whereas least among 0–15 years in July. The susceptibility to infection in people over 70 years was more than those under 20.27 Further, younger/middle-aged adults, children aged under 10 years have significantly lower susceptibility to SARS-CoV-2 infection, while adults aged over 60 years have a high susceptibility to infection.28 Another study found an inverse relationship between age and relative enhancement in the risk of mortality with the Delta variant. Hence, the younger age groups showed a greater increase in their relative risk of death than the older age groups.29
The B.1.351 variant first identified in the early October of 2020 in South Africa30 and P.1 variant identified in Brazilian travelers in January 2021,31 Japan are variants of concern designated by the WHO. The B.1.351 variant has a mutation in the RBD of spike protein, namely, K417N, E484K, and N501Y and the P.1 variant has a mutation in the spike RBD, namely, K417T, E484K, and N501Y. These variants are reported to have an increased risk of transmission and reduced neutralization by monoclonal antibody therapy, convalescent sera, and post-vaccination sera.32,33 We found the prevalence of B.1.351 (1.3%) and P.1 (2.7%) VOCs lower than B.1.1.7 and B.1.617.2 although they raised concerns about potential increased transmission or a propensity for re-infection.
The B.1.427/B.1.429 variants increased transmissibility up to 24% and L452R spike mutation is associated with spike-ACE2 receptor interaction.34 Our data showing B.1.427/B.1.429 and B.1.526 lower than other variants was supported by the CDC, who deescalated Epsilon B.1.427/B.1.429 from a VOC on June 29, 2021 due to significant decrease in the proportion of B.1.427/B.1.429 (Epsilon) variants and effectiveness of vaccines and treatments against it. 1
Strategies for Surveillance and Control of COVID Infection
For using pre-optimized TaqMan assays, ready-to-use master mix and predefined software for analysis can be a convenient tool for screening SARS-CoV-2 mutation in a shorter turnaround time. Although detecting only the predefined mutation panel and failing to detect emerging variants, rapid results and low resources considered it a valuable tool. Also, the present method can select the samples for sequencing for confirmation of any new variants. This method is helpful in low-resource places and countries for the surveillance of VOC and VOI where sequencing is unavailable. WGS is not available to conduct for all samples even in the developed country including the US due to which tracking of COVID-19 infection is getting difficult.35 In a study from January 2021, the majority of B.1.1.7 genomes were identified based on S-gene target failures (SGTF) in community-based diagnostic PCR testing.36 It may be one of the reasons for rising cases of COVID-19 and being difficult to control. Hence, low resource and easily available methods such as genotyping should be used for identifying the existing mutation in the community. It is better to use sequencing only to confirm the identity of certain variants and for continuous surveillance of emerging variants to accelerate the control policies for COVID-19 infection.
Conclusions
TaqMan SARS-CoV-2 mutation panel molecular genotyping assays can be used for detection of these known variants B.1.617.2 (Delta), B.1.1.7 (Alpha), B.1.526 (Iota), B.1.351 (Beta), P.1 (Gamma), P.2 (Zeta), B.1.617.1 (Kappa) and B.1.427/B.1.429 (Epsilon) from patient specimens within a couple of hours. Detection of these variants can assist in surveillance and control of the pandemic as well as in the management of patient infection.
Acknowledgment
We are grateful to the laboratory staff and management of Patient’s Choice Laboratories for their immense support during the study. We acknowledge Mr. Tyler McKee for checking the English in this manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Centers for Disease Control and Prevention [homepage on the Internet]. COVID-19: SARS-CoV-2 variant classifications and definitions. Available from:https://www.cdc.gov/coronavirus/2019-ncov/cases-updates/variant-surveillance/variant-info.html. Accessed August 6, 2021.
2. U.S Food and Drug Administration [homepage on the Internet]. SARS-CoV-2 viral mutations: impact on COVID-19 tests 2021. Available from: https://www.fda.gov/medical-devices/coronavirus-covid-19-and-medical-devices/sars-cov-2-viral-mutations-impact-covid-19-tests. Accessed August 6, 2021.
3. Korber B, Fischer WM, Gnanakaran S, et al. Tracking changes in SARS-CoV-2 spike: evidence that D614G increases infectivity of the COVID-19 virus. Cell. 2020;182(4):812–827.e19. doi:10.1016/j.cell.2020.06.043 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
4. Paul P, France AM, Aoki Y, et al. Genomic surveillance for SARS-CoV-2 variants circulating in the United States, December 2020–May 2021. MMWR Morb Mortal Wkly Rep. 2021;70:846–850. doi:10.15585/mmwr.mm7023a3 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
5. Lind A, Barlinn R, Landaas ET, et al. Rapid SARS-CoV-2 variant monitoring using PCR confirmed by whole genome sequencing in a high-volume diagnostic laboratory. J Clin Virol. 2021;141:104906. doi:10.1016/j.jcv.2021.104906 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
6. TaqManTM SARS-CoV-2 mutation panel user guide (package insert). Publication number MAN0024768, 2021.
7. Université de Genève and Hôpitaux Universitaires de Genève. Protocol for specific RT-PCRs for marker regions of the spike region indicative of the UK SARS-CoV-2 variant B.1.1.7 and the South African variant 501Y.V2 2021. Available from: https://www.hug.ch/sites/interhug/files/structures/laboratoire_de_virologie/protocol_amplification_voc_20201201_uk_geneva.pdf. Accessed October 14, 2021.
8. Gulay K, Kolukirik M, Bayrakdar F, et al. 40 minutes RT-qPCR assay for screening spike N501Y and HV69-70del mutations 2021. Available from: https://www.biorxiv.org/content/10.1101/2021.01.26.428302v1.full.pdf. Accessed October 14, 2021.
9. Vogels CBF AT, Breban M, Fauver JR, et al. Multiplexed RT-qPCR to screen for SARS-COV-2 B.1.1.7 variants: preliminary results 2021. Available from: https://virological.org/t/multiplexed-rt-qpcr-to-screen-for-sars-cov-2-b-1-1-7-variants-preliminary-results/588. Accessed October 14, 2021.
10. Public Health England. SARS-CoV-2 lateral flow antigen tests: evaluation of VUI-202012/01 2020. [homepage on the internet]. Available from: https://www.gov.uk/government/publications/sars-cov-2-lateral-flow-antigen-tests-evaluation-of-vui-20201201/sars-cov-2-lateral-flow-antigen-tests-evaluation-of-vui-20201201. Accessed October 14, 2021.
11. Hirotsu Y, Omata M. Detection of R.1 lineage severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) with spike protein W152L/ E484K/G769V mutations in Japan. PLoS Pathog. 2021;17(6):e1009619. doi:10.1371/journal.ppat.1009619 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
12. Zhao S, Ran J, Han L. Exploring the interaction between E484K and N501Y substitutions of SARS-CoV-2 in shaping the transmission advantage of COVID-19 in Brazil: a Modeling Study. Am J Trop Med Hyg. 2021;tpmd210412. doi:10.4269/ajtmh.21-0412 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
13. Barton MI, MacGowan SA, Kutuzov MA, Dushek O, Barton GJ, van der Merwe PA. Effects of common mutations in the SARS-CoV-2 spike RBD and its ligand, the human ACE2 receptor on binding affinity and kinetics. Elife. 2021;10:e70658. PMID: 34435953. doi:10.7554/eLife.70658 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
14. Hu J, Li C, Wang S, Li T, Zhang H. Genetic variants are identified to increase risk of COVID-19 related mortality from UK biobank data. medRxiv. 2020:20226761. PMID: 33200144. doi:10.1101/2020.11.05.20226761 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
15. Pivonello R, Auriemma RS, Pivonello C, et al. Sex disparities in COVID-19 severity and outcome: are men weaker or women stronger? Neuroendocrinology. 2020:1–20. PMID: 33242856. doi:10.1159/000513346 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
16. Yanez ND, Weiss NS, Romand JA, Treggiari MM. COVID-19 mortality risk for older men and women. BMC Public Health. 2020;20(1):1742. PMID: 33213391. doi:10.1186/s12889-020-09826-8 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
17. Pinto BGG, Oliveira AER, Singh Y, et al. ACE2 expression is increased in the lungs of patients with comorbidities associated with severe COVID-19. J Infect Dis. 2020;222(4):556–563. doi:10.1093/infdis/jiaa332 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
18. Isabel S, Graña-Miraglia L, Gutierrez JM, et al. Evolutionary and structural analyses of SARS-CoV-2 D614G spike protein mutation now documented worldwide. Sci Rep. 2020;10(1):14031. PMID: 32820179. doi:10.1038/s41598-020-70827z [PMC free article] [PubMed] [CrossRef] [Google Scholar]
19. Hodcroft EB, Zuber M, Nadeau S, et al. Spread of a SARS-CoV-2 variant through Europe in the summer of 2020. Nature. 2021;595(7869):707–712. doi:10.1038/s41586-021-03677-y [PubMed] [CrossRef] [Google Scholar]
20. Edmunds WJ, Jewell NP, Diaz-Ordaz K, Keogh RH. Increased mortality in community-tested cases of SARS-CoV-2 lineage B.1.1.7. Davies NG, Jarvis CI; CMMID COVID-19 Working Group. Nature. 2021;593(7858):270–274. PMID: 33723411. doi:10.1038/s41586-021-03426-1 [PubMed] [CrossRef] [Google Scholar]
21. Winger A, Thomas C. The spike of concern—the novel variants of SARS-CoV-2. Viruses. 2021;13(6):1002. doi:10.3390/v13061002 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
22. Centers for disease control and prevention [homepage on the internet]. Available from: https://www.cdc.gov/coronavirus/2019-ncov/cases-updates/variantproportions.html:VariantProportions. Accessed October 14, 2021.
23. Peng K, Wang Q, Yin Y, et al. Delta spike P681R mutation enhances SARS-CoV-2 fitness over alpha variant. mSystems. 2021:e0070221. PMID: 34609171. doi:10.1128/mSystems.00702-21 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
24. Li B, Deng A, Li K, et al. Viral infection and transmission in a large, well-traced outbreak caused by the SARS-CoV-2 Delta variant. medRxiv. 2021. doi:10.1101/2021.07.07.21260122 [CrossRef] [Google Scholar]
25. Latif AA, Julia L, Mulle LJ, et al. And the center for viral systems biology. “B.1.1.7 lineage report.” outbreak.info. Available from: https://outbreak.info/situation-reports?pango=B.1.1.7&loc=GBR&loc=USA&loc=USA_US-CA&selected=GBR). Accessed October 14, 2021.
26. Sheikh A, McMenamin J, Taylor B, et al. SARS-CoV-2 delta VOC in Scotland: demographics, risk of hospital admission, and vaccine effectiveness. Lancet. 2021;397(10293):2461–2462. doi:10.1016/S0140-6736(21)01358-1 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
27. Davies NG, Klepac P, Liu Y, et al; CMMID COVID-19 working group. Age-dependent effects in the transmission and control of COVID-19 epidemics. Nat Med. 2020;26(8):1205–1211. doi:10.1038/s41591-020-0962-9 [PubMed] [CrossRef] [Google Scholar]
28. Goldstein E, Lipsitch M, Cevik M. On the effect of age on the transmission of SARS-CoV-2 in households, schools, and the community. J Infect Dis. 2021;223(3):362–369. doi:10.1093/infdis/jiaa691 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
29. Fishman DN, Tuite AR. Age-specific changes in virulence associated with SARS-C0V-2 variants of concern. medRxiv. 2021;09:25.21264097. doi:10.1101/2021.09.25.21264097 [CrossRef] [Google Scholar]
30. Mwenda M, Saasa N, Sinyange N, et al. Detection of B.1.351 SARS-CoV-2 variant strain - Zambia, December 2020. MMWR Morb Mortal Wkly Rep. 2021;70(8):280–282. doi:10.15585/mmwr.mm7008e2 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
31. Fujino T, Nomoto H, Kutsuna S, et al. Novel SARS-CoV-2 variant identified in travelers from Brazil to Japan. Emerg Infect Dis. 2021. doi:10.3201/eid2704.210138 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
32. Ho D, Wang P, Liu L, et al. Increased resistance of SARS-CoV-2 variants B.1.351 and B.1.1.7 to antibody neutralization. Res Sq. 2021:rs.3.rs–155394. PMID: 33532763. doi:10.21203/rs.3.rs-155394/v1 [CrossRef] [Google Scholar]
33. Wang P, Casner RG, Nair MS, et al. Increased resistance of SARS-CoV-2 variant P.1 to antibody neutralization. bioRxiv. 2021:433466. PMID: 33688656. doi:10.1101/2021.03.01.433466 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
34. Deng X, Garcia-Knight MA, Khalid MM, et al. Transmission, infectivity, and neutralization of a spike L452R SARS-CoV-2 variant. Cell. 2021;184(13):3426–3437.e8. PMID: 33991487. doi:10.1016/j.cell.2021.04.025 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
35. Maxmen A. Why US coronavirus tracking can’t keep up with concerning variants. Nature. 2021;592(7854):336–337. doi:10.1038/d41586-021-00908-0 [PubMed] [CrossRef] [Google Scholar]
36. Washington NL, Gangavarapu K, Zeller M, et al. Emergence and rapid transmission of SARS-CoV-2 B.1.1.7 in the United States. Cell. 2021;184(10):2587–2594.e7. doi:10.1016/j.cell.2021.03.052 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
Articles from Infection and Drug Resistance are provided here courtesy of Dove Press

Comments